Publications
Highlights
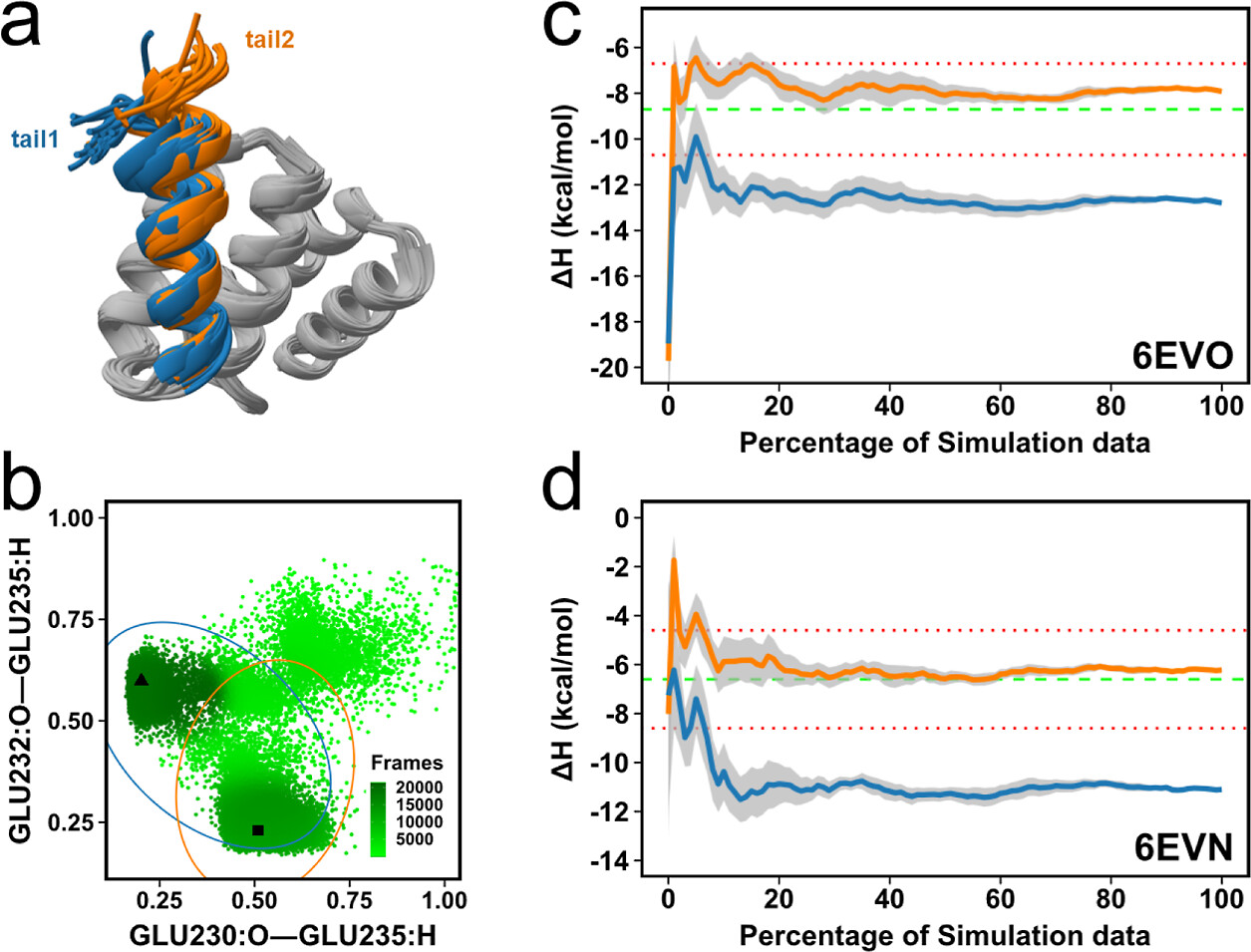
We carefully assembled and curated a set of 11 protein–peptide complexes where there is structural and isothermal titration calorimetry data available and then computed the absolute enthalpy of binding.
Süleyman Selim Çınaroğlu, Philip C Biggin
J. Chem. Inf. Model. 2023, 63, 19, 6095–6108
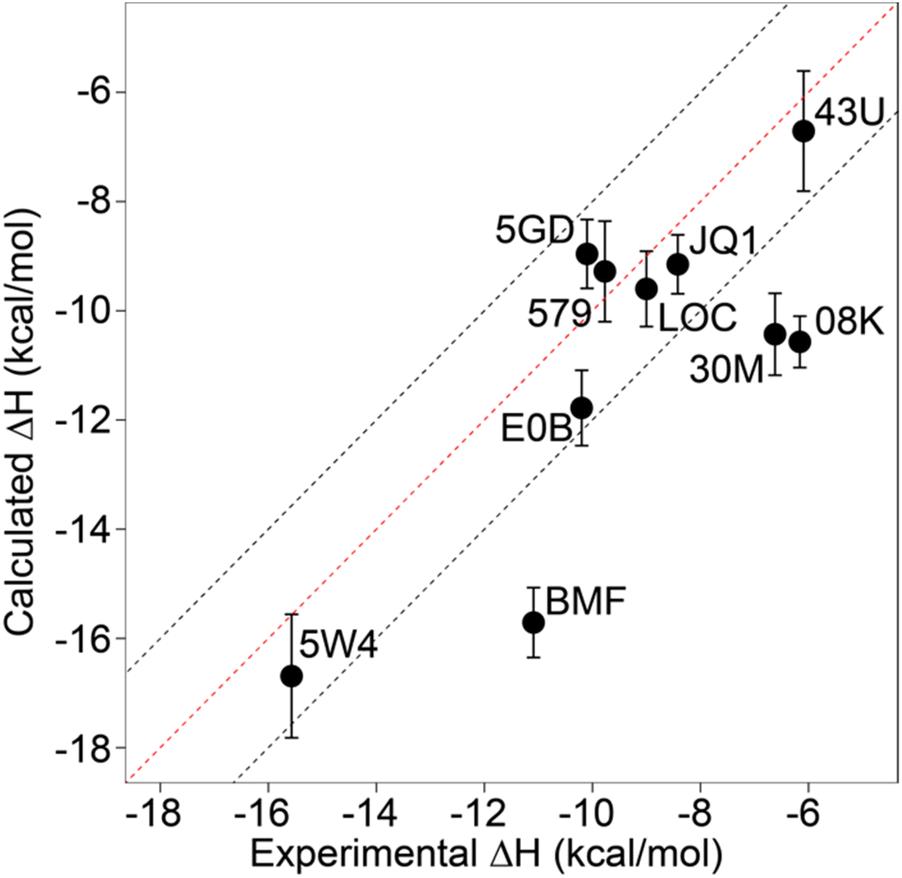
We evaluated the performance of absolute enthalpy of binding calculations using molecular dynamics simulation for ten inhibitors against a member of the bromodomain family, BRD4-1, against isothermal titration calorimetry data.
Süleyman Selim Çınaroğlu, Philip C Biggin
Chem. Sci., 2023, 14, 6792-6805
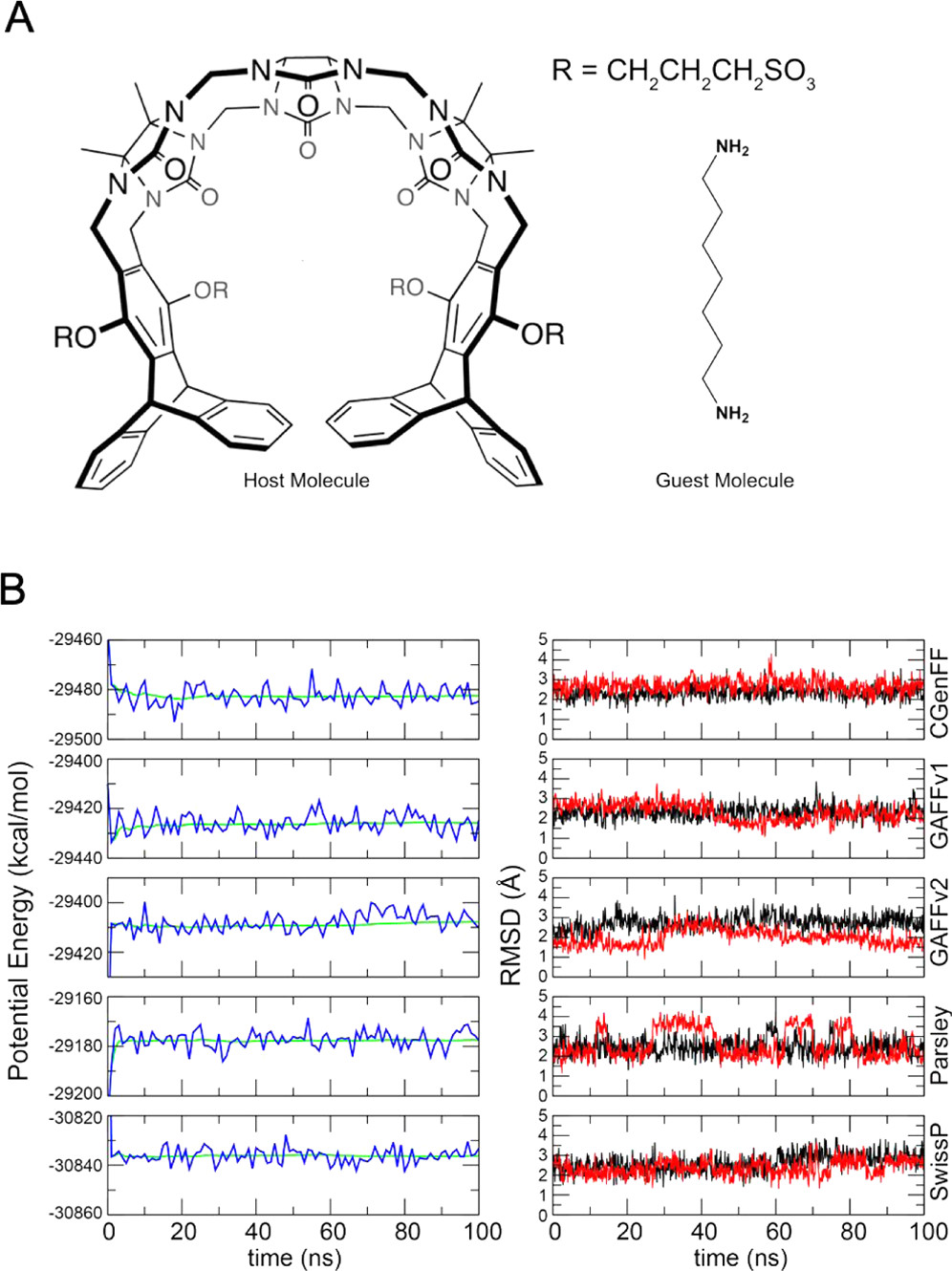
We present absolute enthalpy calculations using the multibox approach on a set of 25 cucurbit[7]uril–guest pairs. Eight water models were considered (TIP3P, TIP4P, TIP4P-Ew, SPC, SPC/E, OPC, TIP5P, Bind3P), along with five force fields commonly used in the literature (GAFFv1, GAFFv2, CGenFF, Parsley, and SwissParam).
Süleyman Selim Çınaroğlu, Philip C Biggin
J. Phys. Chem. B 2021, 125, 6, 1558–1567
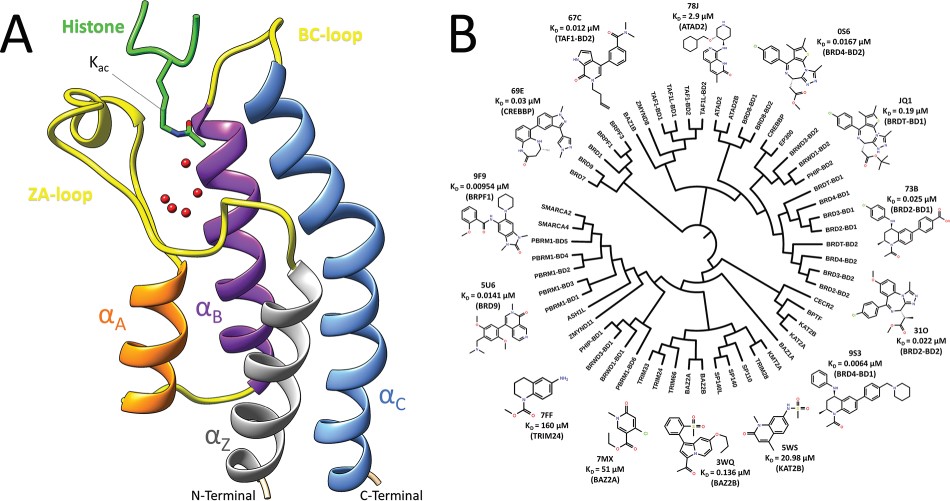
This study reports an extensive testing of different MM-GBSA scoring schemes on two bromodomain (BRD) datasets. The first set is composed of 24 BRPF1 complexes, and the second one is a nonredundant set constructed from the PDBbind and composed of 28 diverse BRD complexes. A variety of MM-GBSA schemes were analyzed to evaluate the performance of four protocols with different numbers of minimization and MD steps, 10 different force fields and three different water models.
Süleyman Selim Çınaroğlu, Emel Timuçin
Briefings in Bioinformatics, 21(6), 2020, 2112–2125
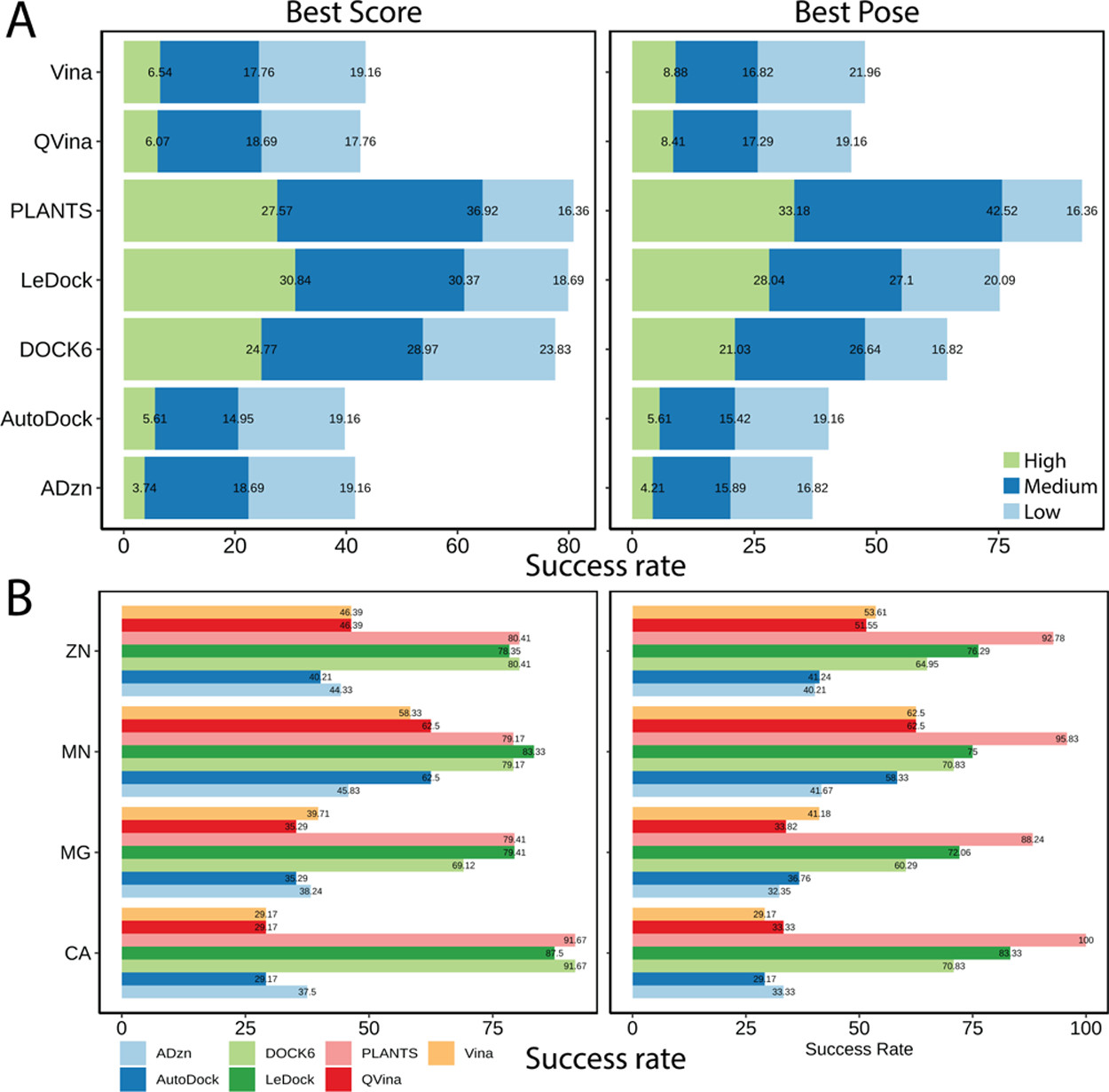
This study reports comparative assessment of seven docking programs (AutoDock4, AutoDock4Zn, AutoDock Vina, Quick Vina 2, LeDock, PLANTS, and UCSF DOCK6) on a diverse metalloprotein set which was compiled for this study. The refined set of the PDBbind (2017) was screened to gather 710 complexes with metal ion(s) closely located to the ligands (<4 Å).
Süleyman Selim Çınaroğlu, Emel Timuçin
J. Chem. Inf. Model. 2019, 59, 9, 3846–3859
Full List
For a full list, go to Google Scholar, Web of Science