Research in Molecular Modeling & Drug Discovery
Our lab harnesses the power of computational tools to unravel the mysteries of biomolecular dynamics and accelerate the discovery of therapeutic agents.
Biomolecular Structure and Dynamics
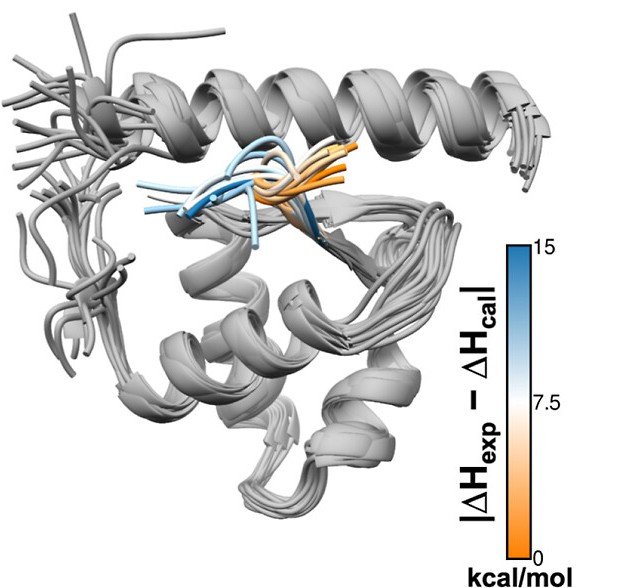
Utilizing molecular dynamics simulations, we explore the intricate organization of biomolecules and macromolecular complexes and the interactions that define biological systems at the molecular level. By employing sophisticated simulation techniques, we create comprehensive models that capture these dynamics over time scales from picoseconds to milliseconds. These detailed models are instrumental in investigating how external factors, such as the presence of ligands or the impact of genetic mutations, influence biomolecular behaviour. Our research focuses on decoding the complex processes of structural dynamics that are fundamental to cellular signalling and regulatory mechanisms.
By leveraging the power of high-performance computational simulations, extensive database analyses, and statistical mechanics principles, we aim to elucidate the atomistic details of biomolecular motions. Understanding these principles helps us to dissect the underlying mechanisms that govern the function and dynamics of biomolecules. We are dedicated to solving pressing health challenges, including fighting antibiotic resistance and developing more effective treatments for neurodegenerative conditions, cancer, and viral diseases. Our goal is to transform fundamental scientific discoveries into actionable medical solutions.
-
Çınaroğlu, S.S., & Biggin, P.C. (2023). Journal of Chemical Information and Modeling, 63(19), 6095-6108.
-
Çınaroğlu, S.S., & Biggin, P.C. (2023). Chemical Science, 14(24), 6792-6805.
Computer-Aided Drug Discovery
We leverage advanced computational techniques to enhance the drug discovery process. Our work integrates molecular modelling, dynamic simulations, and data analytics to accelerate the identification and optimization of new therapeutic agents. We aim to build drug discovery pipelines by developing proprietary algorithms and models that increase the speed and reduce the costs of bringing new treatments to market.
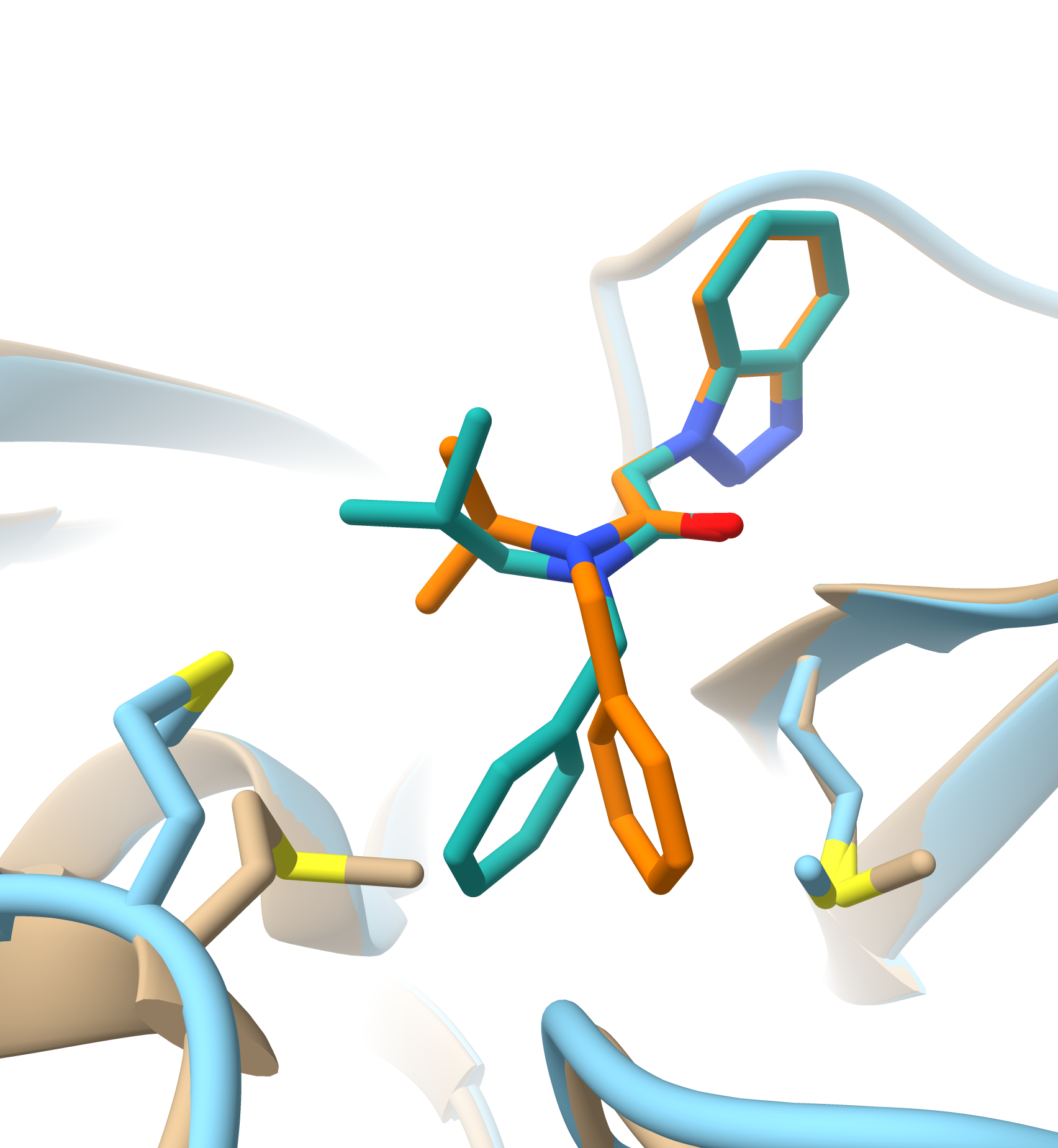
A core focus of our work is the application of structure-based drug design, leveraging protein-ligand interaction studies to inform the development of therapeutic agents. By integrating quantitative structure-activity relationship (QSAR) models and bioinformatics, we enhance our understanding of how molecular structures correlate with biological activity, which is crucial for designing more targeted and effective therapies.
-
Çınaroğlu, S.S., & Timuçin, E. (2019). Journal of Chemical Information and Modeling, 59(9), 3846-3859.
-
Çınaroğlu, S.S., & Timuçin, E. (2020). Briefings in Bioinformatics, 21(6), 2112-2125.
Host-Guest Chemistry
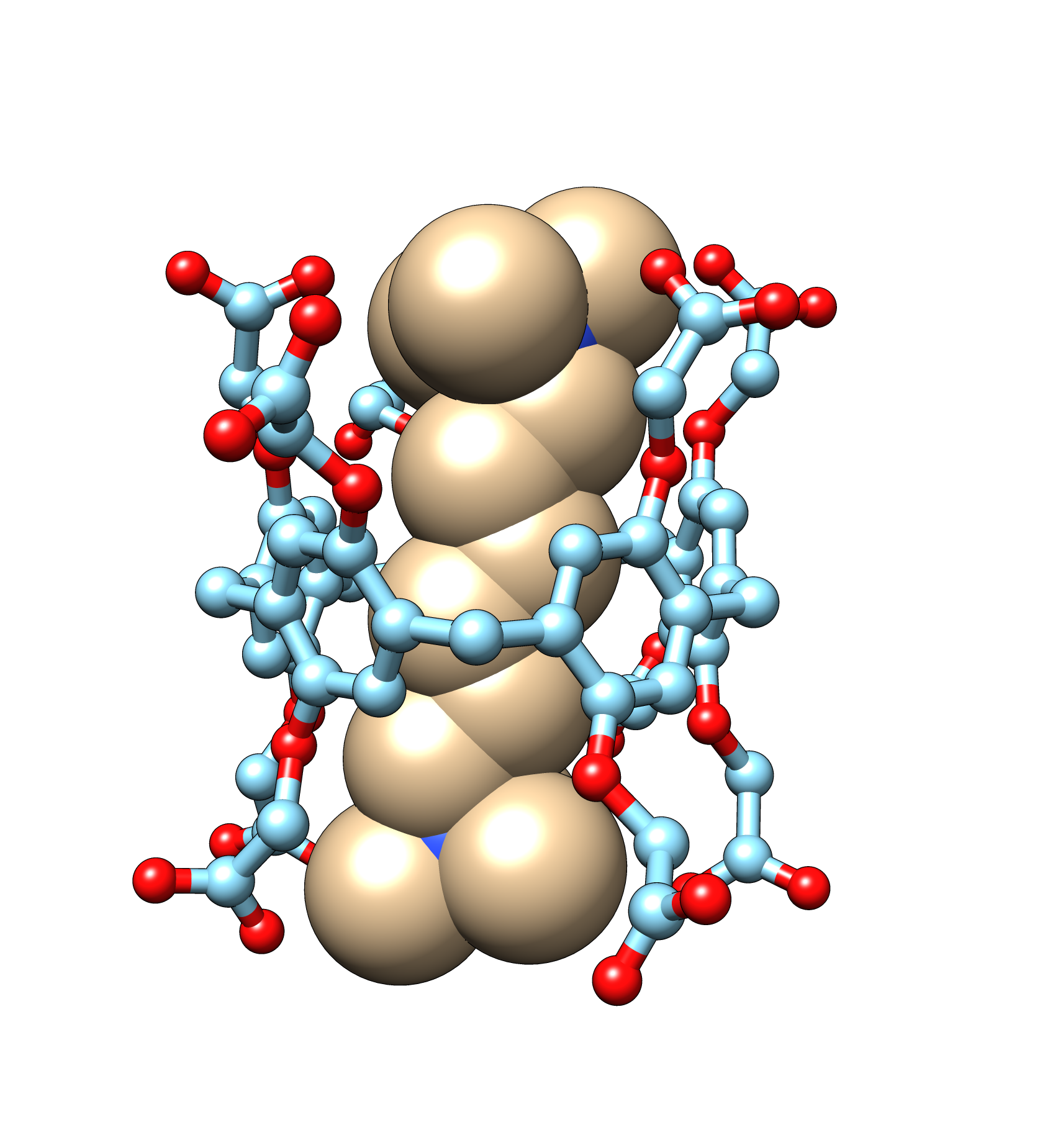
Host-guest systems emerge as pivotal models in our quest for precision in molecular dynamics (MD) simulations. They mirror the complexities of larger protein-ligand systems but demand far less computational power. Their binding affinities align closely with those observed in protein-ligand interactions, establishing them as ideal subjects for detailed molecular recognition studies.
Our research group leverages host-guest systems’ simplicity and efficiency to enhance the reliability of molecular dynamics simulations. Using these systems, we aim to advance our understanding of molecular interactions, ultimately improving the accuracy of our simulations across more demanding biological systems. This foundational work is crucial for pushing the boundaries of what’s possible in computational molecular modelling.
-
Çınaroğlu, S.S., & Biggin, P.C. (2021). The Journal of Physical Chemistry B, 125(6), 1558-1567.
VirtualFlow Türkiye

Our lab actively contributes to the VirtualFlow project, led by Dr. Christoph Gorgulla. VirtualFlow is a free, open-source initiative to facilitate widespread use and dynamic development within the scientific community. It encourages researchers to join the project and contribute to enhancing existing features and expanding its functionality. For more information and resources, please visit the official VirtualFlow homepage at https://www.virtual-flow.org.
A key component of VirtualFlow is the VirtualFlow virtual screen (VFVS), a powerful tool designed to explore vast regions of chemical space in search of promising small-molecule binders. VFVS leverages high-performance computing resources, which continue to grow in availability and power, and innovative virtual screening databases like the Chemical Universe Databases, which house billions to trillions of compounds yet to be fully explored.
-
Gorgulla, C., Çınaroğlu, S.S., Fischer, P. D., Fackeldey, K., Wagner, G., & Arthanari, H. (2021). International Journal of Molecular Sciences, 22(11), 5807.
-
Gorgulla, C., Nigam, A., Koop, M., Çınaroğlu, S.S., S., Secker, C., Haddadnia, M., … & Arthanari, H. (2023). bioRxiv, 2023-04.
Our research highlights are summarized in the publications listed at Publications